

A doença de Huntington também é conhecida como Coreia de Huntington e possui esse nome em homenagem ao autor da primeira publicação científica da síndrome clínica, que foi apresentada em abril de 1872, George Huntington.
O termo Coreia vem de origem grega e significa dança, sendo uma designação muito apropriada para as alterações motoras presentes nesta síndrome, semelhantes a alguns passos de dança.
Segundo o artigo original, George Huntington descreveu uma série de alterações, em que a característica mais marcante e típica é um espasmo clônico afetando os músculos voluntários sem ocorrer perda da sensibilidade ou consciência.
Segundo Huntington:
“A doença comumente se inicia por leves abalos dos músculos da face, que aumenta gradativamente em violência e variedade. As pálpebras são mantidas piscando, a testa franzida depois elevada, o nariz torcido para um lado e depois para o outro e a boca se volta em direções variadas, dando o paciente a aparência mais ridícula que se possa imaginar. Parece haver alguma força oculta, algo que está de certa forma brincando com a vontade e de algum modo dificultando e pervertendo seus desígnios; e depois que a vontade para de exercer sua força numa direção qualquer, assume o controle e mantém a pobre vítima numa agitação contínua enquanto ela permanece acordada”.
A prevalência de indivíduos afetados com doença de Huntington tem uma distribuição relativamente homogênea em todo mundo, variando de 5 a 10 casos por 100.000 indivíduos. No Brasil não existem dados sobre a real prevalência ou incidência da coreia de Huntington.
Desenvolvimento da doença de Huntington
As coreias podem ter causas genéticas ou não-genéticas. Coreias vasculares, autoimunes, metabólicas, tóxicas e induzidas por medicamentos são exemplos de coreias não-genéticas.
As coreias genéticas são geralmente diagnosticadas com base em testes genéticos e podem ter origem autossômica, dominante ou recessiva, ou em cromossomas sexuais.
A doença de Huntington tem uma causa única em todos os pacientes, o que permite aos investigadores concentrarem-se em um único mecanismo fundamental da doença. Em 1993 foi identificada a localização do gene responsável pela doença.
Causa genética
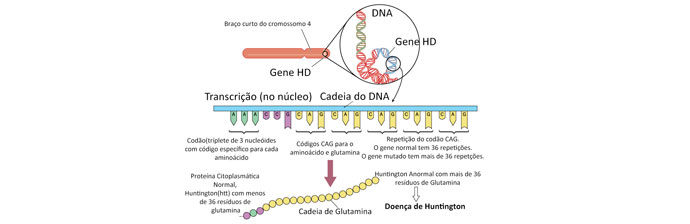
Todos os indivíduos têm duas cópias do gene Huntingtina (HTT), que codifica a proteína Huntingtina (Htt). O gene também é chamado “gene HD” e IT15 (do inglês interesting transcripit 15’). Parte deste gene consiste em repetições de trinucleotídeos, que, em extensão, variam entre indivíduos e podem variar entre gerações.
Quando a extensão desta seção de repetições atinge um determinado comprimento, é produzida uma proteína de forma alterada, chama de proteína de Huntingtina mutante (mHtt). Esta diferente forma proteica apresenta funções diferentes da proteína não mutada, sendo assim responsável pela patogenia da patologia. A mutação produzida é geneticamente dominante e com penetrância quase completa, o que significa que a herança do gene mutado de qualquer progenitor causa a doença.
Não está ligada ao sexo, mas sim ao comprimento da seção repetida do gene, sendo que a sua gravidade pode ser influenciada pelo sexo do progenitor afetado. Como existe maior instabilidade da espermatogênese do que na oogênese, as maiores repetições podem ocorrer no sexo masculino.
Mutação genética
A doença de Huntington é uma das doenças associadas à repetição de trinucleotídeos, causada por um excessivo comprimento da seção repetida. O gene do HTT está localizado no braço curto do cromossomo 4 e contém uma sequência de bases de ADN citosina-adenina-guanina (CAG), repetidas múltiplas vezes.
A sequência CAG codifica o aminoácido “glutamina”, logo a repetição CAG resulta na produção de uma cadeia de glutamina conhecida como “poliglutamin tract” (ou poliQ tract), e a parte do gene repetida como “região poliQ”.
Um indivíduo normalmente tem menos que 36 repetições de glutamina na região poliQ que resulta na produção da proteína citoplasmática Huntingtina. No entanto, sequências de 36 ou mais glutaminas resultam na produção de uma proteína com características ligeiramente diferentes. Repetições entre 36 e 39 resultam numa forma de penetrância incompleta, com início de sintomas mais tardios e progressão lenta. Em alguns casos, o início pode ser tão tardio que os doentes não chegam a ter sintomas.
Quando existe mais de 40 repetições CAG, a doença de Huntington apresenta penetrância completa e pode ocorrer antes dos 20 anos, sendo chamada de doença de Huntington juvenil, “acinética-rígida” ou variante de Westphal.
O número de repetições CAG está relacionado com a extensão do dano, explicando a variação da idade de início dos sintomas em cerca de 40% dos doentes. O restante é atribuído ao ambiente e outros genes que modificam os mecanismos da doença de Huntington.
Repetições mais longas resultam em início mais precoce dos sintomas e maior rapidez na progressão.
Transmissão
A Doença de Huntington tem herança autossômica dominante, o que significa que um indivíduo afetado herda uma cópia do gene mutado de um progenitor afetado.
Uma vez que a penetrância do gene é muito alta, os indivíduos que possuem o gene mutado terão a doença. Neste tipo padrão de transmissão, cada filho tem 50% de probabilidade de herdar o gene mutado e ter a doença. É independente do sexo e o fenótipo não salta gerações.
Mecanismo
A proteína Huntingtina interage com mais de 100 proteínas e parece ter múltiplas funções biológicas. O comportamento da mHtt não é totalmente compreendido, mas sabe-se que é tóxico para vários tecidos, principalmente o cérebro.
Os danos precoces são mais evidentes do striatum, mas à medida que a doença progride, outras áreas do cérebro também são afetadas.
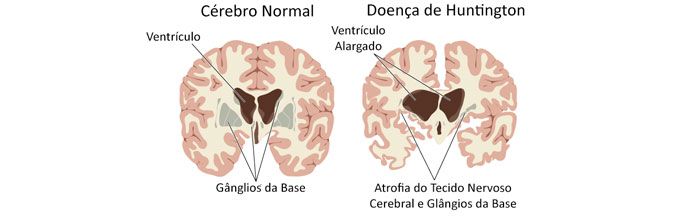
Alterações macroscópicas
A Doença de Huntington afeta todo o cérebro, mas existem áreas mais vulneráveis que outras.
Os primeiros efeitos são mais evidentes dos gânglios da base, em especial o striatum, que é composto pelo núcleo caudado e putâmen. O striatum é a principal estrutura de input nos gânglios da base, a partir do córtex cerebral, tálamo e tronco cerebral. Os seus neurônios se projetam para o globo pálido e substância negra. Essas duas estruturas, em conjunto, são os principais locais de output dos gânglios da base.
Além dessas áreas, a substância negra, as camadas, 3, 5 e 6 do córtex cerebral, o hipocampo, as células de purkinje do cerebelo, núcleo lateral tuberal do hipotálamo e partes do tálamo também são afetadas.
Essas áreas são afetadas de acordo com sua estrutura e o tipo de neurônios que contém, diminuindo o seu tamanho à medida que vão perdendo células. A doença também causa o aumento anormal de astrócitos e ativação das células imunitárias cerebrais, a micróglia.
Fatores de risco
Não existem fatores de risco conhecidos para a Doença de Huntington, já que é uma doença de origem genética.
Sintomas
A Doença de Huntington é um distúrbio degenerativo progressivo que causa alterações no controle motor e emocional, prejuízo na habilidade cognitiva e o aparecimento de movimentos involuntários, classicamente a coreia.
A doença no adulto tem geralmente início insidioso de inabilidade e movimentos adventícios, inquietos, aleatórios e rápidos. Sinais clínicos como nistagmo, disartria, movimentos disrítmicos e repetitivos dos dedos o da língua e a presença de reflexos aumentados podem participar do quadro inicial da doença.
-
Distúrbio de movimento
A coreia é o sinal motor mais notável da doença, presente em 90% dos casos. Os movimentos involuntários estão continuamente presentes durante o período em que o paciente está em alerta, sendo o mesmo incapaz de suprimi-los.
As manifestações coreiformes na face são comuns, representadas por contrações na bochecha, ataxia ocular, franzimento das sobrancelhas e movimentos labiais com formações de bico. A fixação do olhar para cima está prejudicada. É comum o envolvimento do pescoço com movimentos anteriores e posteriores e rotação da cabeça.
A respiração pode estar alterada. Os membros inferiores podem ser cruzados e descruzados de forma alternada, e os dedos sofrem movimentos de flexão e extensão.
Geralmente a coreia inicia distalmente, mas conforme a evolução da doença, torna-se generalizada e pode inclusive interromper movimentos voluntários.
A distonia (movimentos lentos anormais e alterações posturais) não é muito frequente no início das manifestações, porém torna-se proeminente no estágio final da enfermidade.
A bradicinesia e a rigidez também não são comuns nas fases iniciais da doença, porém, gradualmente essas manifestações vão aparecendo, dominando o estágio final onde o paciente se torna severamente rígido e acinético.
Um sinal precoce da doença é a incapacidade de realizar corretamente movimentos sequenciais, ou de executar de maneira rápida e harmônica movimentos simples, repetidas vezes. Algumas alterações sutis na marcha podem ser observadas no começo da doença, e com a progressão, as dificuldades tornam-se maiores. Por causa dessas dificuldades, os pacientes apresentam quedas frequentes e a utilização de cadeira de rodas torna-se necessária.
Os pacientes são incapazes de aprender habilidades motoras complicadas e a perda do controle motor progride até o paciente atingir incapacidade de efetuar qualquer movimento proposto.
Inicialmente há um leve distúrbio na pronúncia, o qual é agravado com o tempo. A disfagia é um sintoma que ocorre tardiamente, e a asfixia ou a aspiração decorrentes da disfagia são causas bastante comuns de morbidade entre os doentes.
-
Distúrbio cognitivo
Um declínio da capacidade cognitiva global está presente na maioria dos pacientes com doença de Huntington. Em geral, encontra-se precocemente:
- Lentidão de pensamento;
- Alterações da personalidade;
- Mudanças afetivas;
- Diminuição da capacidade e integrar conhecimentos novos.
A demência é o sintoma inicial em cerca de 10% dos casos e pelo menos 90% dos pacientes desenvolvem demência durante a doença. A dificuldade de executar tarefas simples como permanecer com o olhar fixo lateralmente, manter a protrusão da língua ou fechar firmemente os olhos são manifestações relacionadas tanto à distração experimentada pelos pacientes, como aos distúrbios motores presentes.
A incapacidade de organizar, sequenciar, planejar, coordenar e iniciar movimentos complexos ou manter mentalmente a estratégia motora são manifestações precoces da doença.
-
Distúrbio Psiquiátrico
Menos consistentes que as alterações motoras e cognitivas, os transtornos psiquiátricos também são considerados manifestações clínicas da doença, não estando diretamente relacionados com a severidade da coreia ou demência.
Mudanças no humor e no afeto são comuns, variando desde ansiedade e irritabilidade a longos períodos de depressão. A taxa de suicídio é maior os pacientes de doença de Huntington do que na população em geral. A depressão é o sintoma psiquiátrico mais comum, acometendo cerca de 40% dos pacientes.
Os distúrbios de conduta como apatia, comportamento agressivo, desinibição sexual e alcoolismo podem ser considerados manifestações de declínio cognitivo progressivo como anormalidades relacionados com a alteração de humor.
Outro transtorno psiquiátrico comum na doença de Huntington é a presença de pensamentos ilusórios, que acometem cerca de 50% dos pacientes em estágio avançado. Esses pensamentos podem acompanhar episódios depressivos e maníacos ou podem se manifestar de forma isolada. A psicose é relatada em cerca de 20% dos casos.
Nos estágios mais avançados da doença, pode-se observar insônia e inversão do ritmo circadiano. Os movimentos involuntários desaparecem durante o sono.
Fase terminal
A pneumonia por aspiração é a causa mais comum de morte na fase terminal. A insuficiência cardiorrespiratória e o hematoma subdural (decorrente de um trauma encefálico) são também causas frequentes do óbito.
A duração da doença entre o início e a morte do paciente é de 15 a 20 anos na doença de Huntington do adulto.
Sintomas da doença de Huntington juvenil
A Doença de Huntington Juvenil se caracteriza quando o início dos sintomas surge de forma precoce, afetando pacientes com menos de 20 anos. Essa forma da doença também é conhecida como Variante Westphal ou rígido acinética.
A forma juvenil representa cerca de 5,4% dos casos da doença, e geralmente se manifesta pela primeira vez através de parkisonismo progressivo, demência e convulsões, sendo menos frequente a coreia. A rigidez é o sintoma dominante.
Pacientes adultos com doença de Huntington apresentam crises convulsivas com uma frequência similar ao resto da população (1%), enquanto 30 a 50% dos doentes com a forma juvenil apresentam essas crises.
A Doença de Huntington juvenil possui, em geral, progressão acelerada, diferente dos pacientes que são acometidos tardiamente e apresentam uma evolução branda e arrastada.
A duração da doença entre o início e a morte do paciente na variante juvenil é de 8 a 10 anos.
Diagnóstico
O diagnóstico da doença de Huntington pode ser realizado baseado na presença de manifestações clínicas de disfunção motora progressiva, envolvendo movimentos voluntários e involuntários, acompanhados de distúrbios mentais como déficits cognitivos, distúrbios afetivos e alterações de personalidade em pacientes que apresentam histórico familiar de doença de Huntington.
Exames de imagem
Exames de imagem como a tomografia computadorizada e a ressonância magnética podem ser usados para estudar as alterações neuroanatômicas que ocorrem nos pacientes com doença de Huntington e também para documentar as alterações e correlaciona-las com a evolução clínica da doença, além de fornecer um parâmetro para avaliar algum tratamento proposto.
A alteração encontrada na tomografia computadorizada e na ressonância magnética é a atrofia dos núcleos caudado e putâmen, sem alterar o globo pálido. Essas alterações ocorrem mais rapidamente em pacientes que desenvolvem a doença ainda jovens quando comparados aos que apresentam a doença em uma idade mais avançada.
Se o exame apresenta atrofia do núcleo caudado e putâmem, o diagnóstico da doença de Huntington é fortemente sugerido, apesar de poucas outras afecções apresentarem os mesmos sinais.
O teste genético pode ser usado para confirmar o diagnóstico em caso de ausência de história familiar.
Teste genético pré-sintomático
Caso haja histórico de doença de Huntington na família e o indivíduo não apresente sintomas, ele pode realizar o teste pré-sintomático para saber se possui ou não gene da doença.
O teste pré-sintomático é um evento com elevado peso na vida do indivíduo e uma decisão muito pessoal. Mais de 95% dos indivíduos em risco opta por não realizar o teste, já que não existe cura para a doença.
Geralmente a principal razão para a realização do teste é ajudar em decisões relacionadas com a carreira e a família. Um ponto essencial é ansiedade de não saber se é ou não portador, comparado com o impacto do teste positivo.
A descoberta de que não é afetado pode gerar um efeito chamado “survivor guilt”, que se caracteriza por um sentimento de culpa em relação aos familiares afetados.
O aconselhamento genético pode fornecer informações, conselhos e apoio no momento da decisão de fazer o teste e durante todas as fases de processo do teste, caso o indivíduo escolha realiza-lo.
Nunca se deve forçar um indivíduo a fazer o teste. Crianças em geral não tem discernimento para considerar todas as implicações do teste, por isso a idade mínima requerida para isso costuma ser 21 anos.
O procedimento de teste envolve sessões com vários profissionais.
Exames Neurológicos
O exame neurológico é indicado para assegurar que a pessoa em risco não está demonstrando sintomas da doença e é realmente “pré-sintomático”. É um cuidado necessário para diferenciar um diagnóstico de doença de Huntington a partir de sintomas clínicos e a descoberta que a pessoa é portadora do gene.
O exame neurológico também pode revelar sinais amenos e indicadores da doença de Huntington, mas que não são suficientes para fechar o diagnóstico.
Uma pessoa que apresenta disfunções sutis pode estar em risco de ser portadora do gene ou relativamente propensa a ter mais sintomas patentes.
O exame neurológico pode ajudar a fornecer informações sobre o quanto o paciente precisa ser acompanhando de perto logo após o resultado do teste.
Diagnóstico psicológico e/ou psiquiátrico
O diagnóstico psicológico e psiquiátrico é recomendado considerando os altos níveis de estresse detectados nos indivíduos em risco.
É importante que a avaliação psicológica de estabilidade emocional não seja considerada como um obstáculo a ser ignorado para a qualificação do teste, mas sim como uma forma de identificar pessoas que tem uma maior tendência a precisar de apoio emocional após o teste.
Em casos em que o indivíduo esteja com risco para o suicídio ou possua sintomas maiores de depressão, é apropriado que o teste seja adiado e que um tratamento psiquiátrico seja realizado para que a pessoa fique tranquila antes de passar pelo teste.
Testes Neuropsicológicos
Os testes neuropsicológicos são usados por alguns centros de teste genéticos como parte da avaliação pré-teste.
Muitos centros utilizam os testes neurospsicológicos depois do teste para estabelecer um padrão de comportamento e para poder auxiliar da melhor forma no início da manifestação dos sintomas ou em emergências do problema que podem danificar seriamente a função psicológica.
Abordagem fisioterapêutica
O tratamento fisioterapêutico tem como base a manutenção das habilidades preservadas e a melhora, dentro do possível, das funções já comprometidas.
A intervenção tem como objetivo buscar a promoção da autoconfiança e o aumento da autoestima, tendo como base as necessidades e os desejos do próprio paciente, para motivá-lo a manter sua energia e colaboração.
Para um programa de reabilitação efetivo é importante o trabalho de uma equipe multidisciplinar, além da integração com a família e a comunidade na qual o paciente vive.
Os objetivos da reabilitação devem enfocar a independência das atividades de vida diária e nas atividades profissionais, sem esquecer que durante esse processo, o paciente também necessita de recreação e de atividade esportiva.
O trabalho terapêutico não deve estar focado apenas na fisioterapia convencional, mas também em estratégias de recreação terapêutica, para proporcionar oportunidades de praticar atividades que tragam momentos de prazer e satisfação. A inclusão de música, trabalhos manuais e tarefas em grupo deverá ser implementada sempre que possível, para estabelecer níveis de motivação satisfatórios ao aumento ou manutenção da máxima independência.
Todas as fases do tratamento fisioterapêutico devem ter como objetivo minimizar os efeitos das anormalidades motoras como alterações de tônus, limitações de amplitudes de movimento e instalação de deformidades. Além disso, deve-se prevenir ou tratar possíveis comprometimentos respiratórios.
A intervenção da fisioterapia nos estágios iniciais e intermediários deve focar na manutenção do equilíbrio e da mobilidade, através de exercícios de alongamento para prevenção de contraturas e deformidades e treino para manutenção de equilíbrio com exercícios proprioceptivos.
Nos estágios mais avançados, onde a dependência física se faz mais evidente, o treino de transferência (leito para cadeira de rodas, cadeira para banho, etc.) deve ser enfatizado para se estender ao máximo a independência funcional.
As técnicas fisioterapêuticas utilizadas devem ser específicas ao comprometimento, como em qualquer tipo de doença neurológica, com treino de consciência corporal, controle postural e coordenação motora, com ênfase na realização das atividades de vida diária e manutenção da independência funcional.
Exercícios para o tratamento da doença
A reabilitação da doença de Huntington se assemelha aos tratamentos de reabilitação neurológica. Os exercícios a seguir são indicados para alongamento, fortalecimento e treino de equilíbrio e podem ser realizados nas fases iniciais e intermediárias da doença, quando o paciente ainda possui controle motor dos seus movimentos.
Alongamentos
- Paciente em decúbito dorsal, com um membro flexionado apoiado e o outro em extensão. Passar uma faixa na ponta do pé da perna estendida e solicitar que o paciente segure a faixa, realizando uma flexão de quadril mantendo a extensão de joelho, para alongamento ativo de posteriores de perna.
- Paciente em decúbito dorsal. Solicitar que abrace as duas pernas para alongamento de região glútea e lombar.
Quando o paciente não puder mais realizar os alongamentos sozinhos, o fisioterapeuta deverá fazê-lo de forma passiva.
Fortalecimentos
- Paciente sentado. Solicitar que realize extensão de joelhos para fortalecimento de quadríceps.
- Paciente sentado. Passar uma faixa elástica por volta das pernas, na região dos joelhos e solicitar que realize uma abdução de quadril, para fortalecimento de abdutores.
- Paciente sentado com uma bola entre as pernas, na região dos joelhos. Solicitar que pressione a bola com as pernas para fortalecimento de adutores.
- Paciente sentado, segurando uma barra. Solicitar flexão de ombro para fortalecimento de flexores de ombro.
- Paciente em pé, segurando um halter em cada mão. Solicitar flexão de cotovelo para fortalecimento de bíceps braquial.
Treino de equilíbrio
- Paciente em pé, segurando em uma cadeira. Solicitar que se mantenha em equilíbrio em apenas 1 pé.
- Paciente em pé. Solicite que caminhe apoiando um pé exatamente à frente do outro.
- Paciente em pé, em cima de uma superfície instável (colchonete, por exemplo). Solicitar que se mantenha em equilíbrio em cima dessa superfície, primeiramente com os dois pés e depois com 1 pé só.
O papel do fisioterapeuta
O papel do fisioterapeuta na Doença de Huntington é planejar um programa eficiente de reabilitação, que deverá estar pautado nas características específicas de cada paciente e do meio em que vive, de modo a permitir a manutenção ou progressão dos melhores resultados fisio-funcionais e também emocionais.
É importante que o fisioterapeuta considere todas as possibilidades que possam proporcionar ao paciente melhores chances de promoção funcional a partir de um mesmo espetro de capacidades, enfatizando o trabalho com pontos controles, direcionamento de valores, foco no resultado, metas e objetivos de curto, médio e longo prazo, respeitando o sistema multidisciplinar.
Fornecer apoio aos familiares do paciente também é um papel do fisioterapeuta, tanto em relação à orientação sobre como lidar com algumas situações de estresse diante da evolução dos comprometimentos motores do paciente, quanto ao encaminhamento ao profissional da psicologia, para auxiliar na superação das dificuldades emocionais.
Cuidados e restrições
O fisioterapeuta precisa estar preparado para possíveis situações de crises agudas como a asfixia.
No estágio mais avançado da doença, onde o paciente se encontra acamado, é importante mantê-lo bem posicionado com o uso de colchão “caixa de ovo” e coxins, para prevenção de úlceras de decúbito. É importante repassar essas orientações para os familiares e cuidadores para que o posicionamento se mantenha correto durante todo o dia e que a mudança de decúbito ocorra a cada 2 horas.
Conclusão
A doença de Huntington é uma doença neurodegenerativa progressiva de origem genética, que se caracteriza por apresentar movimentos coreicos, alterações cognitivas e comportamentais, levando a incapacidade total e a morte.
Até o momento não existe cura, apenas tratamentos que ajudam a diminuir os sintomas motores e psiquiátricos da doença. O tratamento da doença de Huntington requer uma equipe multidisciplinar que conta com médicos, fisioterapeutas, enfermeiros, fonoaudiólogos e psicólogos e etc.
A fisioterapia visa manter a funcionalidade do indivíduo dentro de suas limitações, trabalhando para garantir que o paciente se mantenha independente para realização de suas atividades de vida diária pela maior quantidade de tempo possível, tendo como objetivo principal oferecer uma melhor qualidade de vida.

Gostei muito,bem explicado,
Este doutor Huntington poderia ter tido um pouco mais de cuidado com as palavras ao se referir a um portador desta doença.
“dando o paciente a aparência mais ridícula que se possa imaginar…”
Achei forte também, porém, lembrei que existem pessoas de aparência feia, mesmo sem nenhuma doença física ou aparente.
Adorei, usarei na minha apresentação.